substructures of amino acid side chains in protein structures; and
3D base arrangements in RNA structures.
The GrAfSS server integrates the search capabilities of five different algorithms into a single interface. The five algorithms are:
SPRITE − 3D Search for PRotein sITEs
ASSAM − Amino acid Search for Substructures And Motifs
IMAAAGINE − Imagine an Amino Acid Arrangement search enGINE
NASSAM − Nucleic Acid Search for Substructures And Motifs
COGNAC − COnnection tables Graphs for Nucleic ACids
The GrAfSS server provides a one stop platform for the annotation and searching of substructures in both protein and RNA structure coordinate data. The search engine has incorporated individual improvements and updates to the five component search engines. The integrated interface was designed to prevent confusion regarding which particular algorithm is applicable for a specific search requirement.
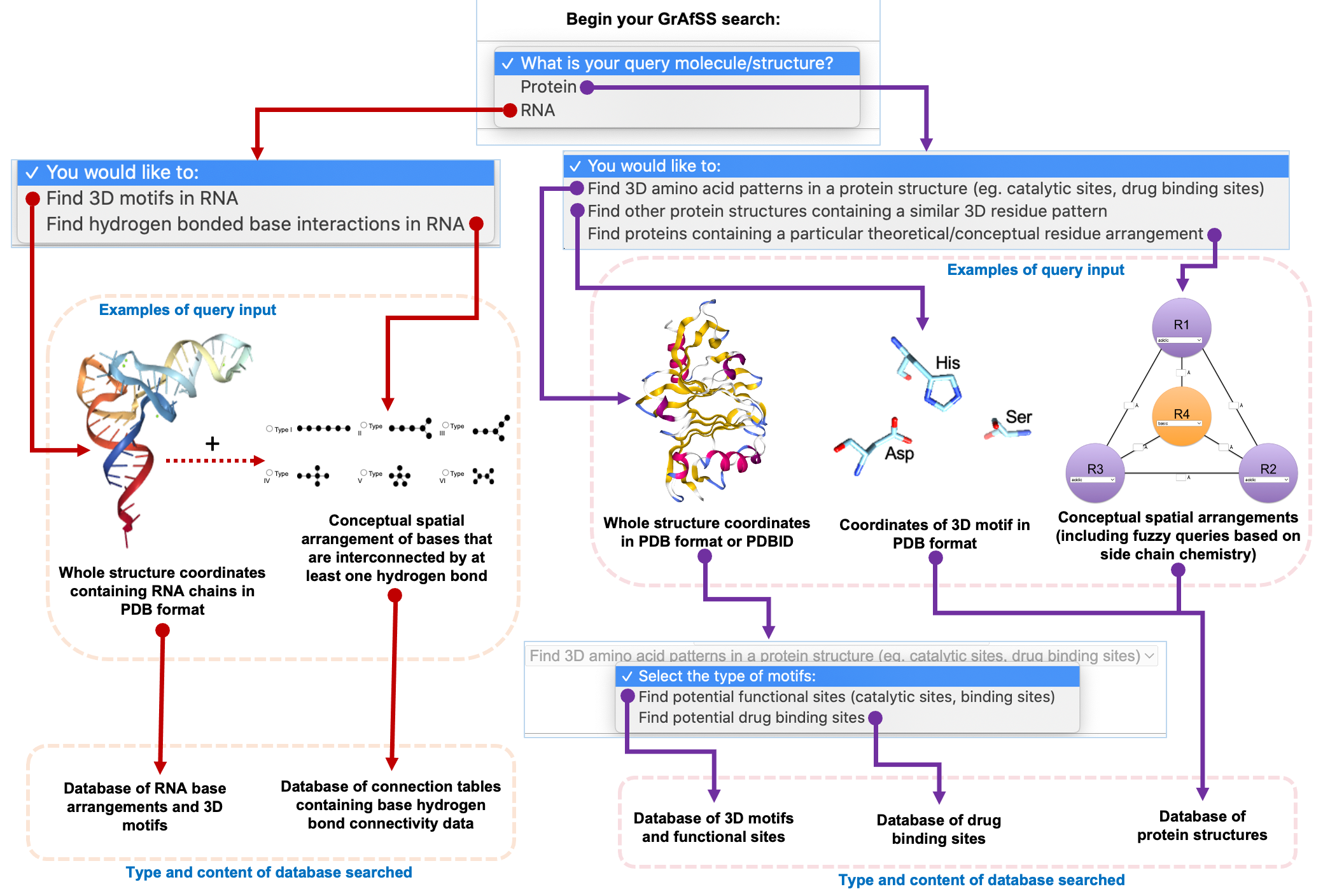
Using the GrAfSS server you can:
Find whether an arrangement of interest, such as a ligand binding site or 3D motif, observed in a protein can be found in other protein structures available in the Protein Data Bank − via the ASSAM algorithm.
Input query: a 3D arrangement or motif (in PDB format) not exceeding 12 residues − for example, a PDB file containing the coordinates of only the serine protease catalytic triad (Asp-Ser-His).
Database: The query is searched against a database of protein structures sourced from the PDB or those that were generated by AlphaFold2 (using the dataset made available by the EBI).
Examples of utility: This search allows users to identify whether a similar 3D side chain arrangement is present in different protein structures, including those that have no fold similarities. Because the search is independent of fold or sequence similarity, it can allow very small functional regions to be identified that cannot otherwise be identified by fold similarity searching. Examples of such motifs include convergently involved catalytic sites. A drug binding site can also be searched against human protein structures to identify whether a drug could have off-target sites that could cause potential side effects or toxicity.
Find whether a hypothetical amino acid arrangement defined by distances between the residues are present protein structures available in the Protein Data Bank or a computationally generated dataset such as the structures predicted by AlphaFold2 (using the dataset made available by the EBI). − via the IMAAAGINE algorithm.
Input query: a hypothetical 3D arrangement of up to 8 residues − for example, a cluster of acidic amino acids that are within a fixed distance constraint to each other.
Database: The query is searched against a database of protein structures sourced from the PDB or those that were generated by AlphaFold2 (using the dataset made available by the EBI).
Examples of utility: This sort of search allows for users to find motifs or 3D arrangements of amino acids that are based on a specific chemistry − such as a region containing a cluster of basic amino acids. It has also been useful in identifying novel motifs that are only detectable in a three-dimensional context, such as a single Histidine from different monomeric subunit that function as an interface for quaternary structure assembly
Find whether known motifs or 3D arrangements of amino acids or RNA bases are present in a query structure coordinate file (protein − using the SPRITE algorithm, and RNA chain containing structures − using the NASSAM algorithm). This would allow for the structural annotation of new structures that were either experimentally or computationally generated (such as by AlphaFold2).
Input query: is a protein structure coordinate file or a structure coordinate file that contains RNA chains.
Database: The query is searched against a database of known motifs such as catalytic sites, binding sites or other motifs that have been curated and added to the database. Examples of other motifs include known drug binding sites.
Examples of utility: The SPRITE and NASSAM searches can annotate a newly solved structure for the presence of known amino acid or base motifs. This can be of use for proteins with uncharacterized functions with no detectable sequence or fold similarity to any examples available in databases such as GenBank, UniProt or PDB. Protein structures that are computationally predicted by AlphaFold2 can also be used as search queries using SPRITE.
Find whether a specific cluster of RNA bases that are connected by hydrogen bonds are present in structure coordinate data containing RNA chains − such as the ribosomal subunits or ribozymes.
Input query: is designed by defining how RNA bases can be interconnected by hydrogen bonds to form a cluster of bases from base pairs to sextuples.
Database: The query is searched against a database of RNA structures sourced from the PDB.
Examples of utility: The COGNAC searches can be used to identify minute conformational differences in RNA structures that are a result of differences in hydrogen bonding. The server can also be used to identify highly stable clusters of bases that are held together in a specific arrangement by hydrogen bonds.
How to start your search:
Identify the type of molecular structure you would like to analyse − a choice of protein structure searching or RNA chain containing structures is available.
Once the type of molecule has been decided, you can then select to satisfy the specific search objective, the respective search interface will then guide you on the next steps to complete the search query.
Once a search has been successfully submitted, a link will be provided to the results for later reference.
The submission progress page will be constantly refreshed until the search has been completed.
A search typically takes within 3-10 minutes depending on the server load.
A search that has been ongoing for more than 2 hours may be terminated by the system − this could be due to either an error or a heavy server load. Terminated searches will need to be repeated.
A search that returns an error could be due to input format problems.
General overview for interpreting your search results:
All GrafSS search outputs provide a listing of the substructural similarities retrieved − this is placed under columns identified as Residues.
For some lists, users have the options of sorting the output depending on specific columns such as structure resolution, or RMSD values.
The RMSD values denote how similar the superpositions for the query are to a match. The lower the RMSD value, the more similar the 3D fit is.
The results also provide information whether the matches retrieve are close to heteroatoms − in many cases, this can be ligands such as metals.
The search outputs allow for users to view the retrieved matches.
The computed structural superpositions are visualized using integrated NGL viewers except for hydrogen bonded base clusters which are visualized using JSMol.
About the search algorithm:
The searches are enabled by representing the atoms of amino acid side chains or RNA bases as peudo-atom vectors. The sub-structural similarities of the pseudo-atom vector representations are then searched using subgraph isomorphism or maximal cliques searching algorithms. This allows the searches to be executed independently of any fold similarity or sequence similarity requirements. Please see the references below for details.
To our knowledge, the integrated search capacity provided by the GrAfSS webserver is not available on any other webserver.
References to earlier working versions of the algorithms implemented as updated versions in the GrAfSS webserver:
Mohd Firdaus-Raih, Hazrina Yusof Hamdani, Nurul Nadzirin, Effirul Ikhwan Ramlan, Peter Willett, Peter J. Artymiuk. 2014. COGNAC: a webserver for searching and annotating hydrogen bonded base interactions in RNA 3-dimensional structures. Nucleic Acids Research, Vol. 42. doi: 10.1093/nar/gku438.
Nurul Nadzirin, Peter Willett, Peter J. Artymiuk, Mohd Firdaus-Raih. 2013. IMAAAGINE: a webserver for the searching of hypothetical 3D amino acid side chain arrangements in the Protein Data Bank. Nucleic Acids Research, Vol. 41: W432-W440. doi: 10.1093/nar/gkt431.
Hazrina Yusof Hamdani, Sri Devan Appasamy, Peter Willett, Peter J. Artymiuk, Mohd Firdaus-Raih. 2012. NASSAM: a server to search for and annotate tertiary interactions and motifs in three-dimensional structures of complex RNA molecules. Nucleic Acids Research, Vol. 40, W35-W41. doi: 10.1093/nar/gks513.
Nurul Nadzirin, Eleanor Gardiner, Peter Willett, Peter J. Artymiuk, Mohd Firdaus-Raih. 2012. SPRITE and ASSAM: web servers for side chain 3D-motif searching in protein structures. Nucleic Acids Research, Vol. 40, W380-W386. doi: 10.1093/nar/gks401.
The GrAfSS server and the associated computer programs are developed and maintained by MFRLab.org at Universiti Kebangsaan Malaysia.
The GrAfSS service is mirrored at:
Malaysia Genome and Vaccine Institute
ISPC, Weizmann Institute of Science (in progress)
Advanced Medical and Institute, Universiti Sains Malaysia (in progress)
Please bookmark or use http://mfrlab.org/grafss and not any IP address displayed in the browser.
For enquiries or to report errors, please email info@mfrlab.org.