
The Drug ReposER webserver is developed using MySQL and PHP scripts through XAMPP application.
Approximately 2% of total PDB structures are found to be co-crystallized with annotated drug molecules (more than 500 PDB Ligand IDs that mapped to drug molecules annotated in the DrugBank database). One protein may bind to more than one drug molecule (e.g. a HIV protease could bind to multiple ligands such as RIT, AB1, 017 and MK1). Vice versa, a single drug molecule can be found in multiple PDB entries (e.g. PDB Ligand ID STR found in multiple PDB entries).
The list of annotated drug molecules was obtained from PDB-drug mappings interface available on the DrugPort database (https://www.ebi.ac.uk/thornton-srv/databases/drugport/). While there is a large collection of drug or drug-like molecules available in the PDB, only those that are annotated as 'approved' drug molecules in the DrugBank database are considered. These drugs include FDA-approved small molecule drugs and FDA-approved biotech drugs.
Nutraceuticals, experimental drugs, solvents (e.g. ethanol [EOH,DB00898]) and glycerol [GOL,DB04077] were excluded.
The Drug ReposER webserver consists of two datasets;
- BINDING_INTERFACES (Dataset of binding interfaces) The dataset consists of all drug binding interfaces derived from the PDB, summing up to 2420 patterns of amino acids containing at least 3 residues, up to 35 residues. Binding residue is defined as any amino acid residue within 4.5Å to any atom of the drug molecule. Binding interfaces with such definition mapped to 2168 representative protein structures (at 100% sequence identity cut-off) bound to total 683 PDB ligand IDs. A large number of binding interfaces bound to solvents, neutraceuticals and experimental drugs were excluded from the list. The structural similarity search engine, the ASSAM program however only search for 3-12 residue containing patterns. Thus these binding sites are converted into multiple fragments of binding residues for the purpose of finding similar patterns in other proteins.
- BINDING_INTERFACES_ASSAM (Dataset of similar patterns of amino acids generated from ASSAM searches) The dataset consists of matched patterns of amino acids similar to drug binding interfaces. The ASSAM program searches for similar patterns of amino acids, given a PDB-formatted pattern of amino acids containing 3-12 amino acid residues. All PDB-formatted patterns of binding interfaces were used for similarity searches, where large binding sites were divided into fragments, due to the limitation of the ASSAM search (Nadzirin et al., 2012) for only 3-12 residue-containing patterns. The searches are based on distances between side chain atoms represented by two pseudoatom vectors, where the program searches for similar substructures in the form of graph representations using distance tolerance of 2.0Å and RMSD cut-off of 1.5Å. In the cases where the ASSAM search returning error, a lower distance tolerance of 1.5Å was used instead. For binding interfaces containing 6 and more binding residues, hits were also retrieved for patterns containing smaller residues, instead of only the exact matches. For example, an ASSAM search of 12-residue binding interface may yield hits that mapped to exact matches of 12-residue patterns, and also matches of 5-residue up to 11-residue containing patterns. Both data sets are interlinked by DrReposER ID.
This interface allow user to identify potential binding interfaces based on local structural similarity searches, either from a query of PDB ID (e.g. '1EQC') or PDB ligand ID (e.g. 'CTS') which returns a list of matches from ASSAM searches. User may know whether the protein of interest could probably have similar patterns of amino acids as known drug binding sites, or the ligand of interest could possibly well-superposed with the drug molecule.
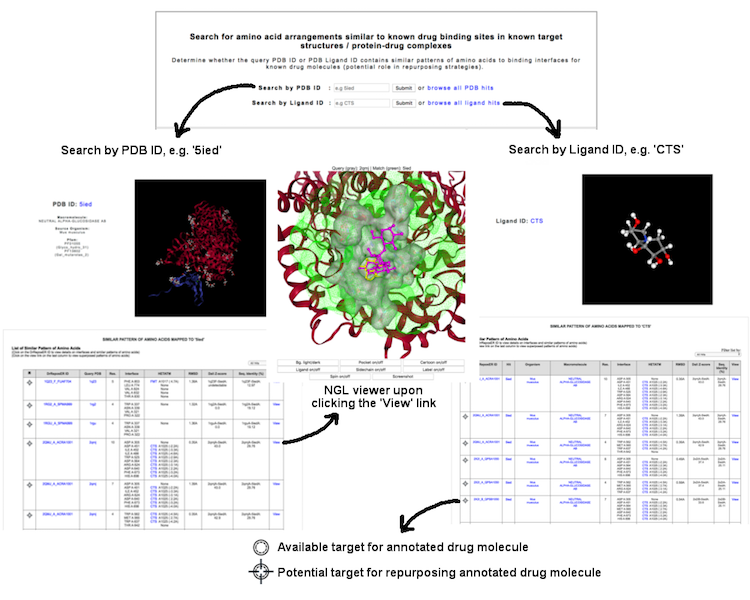
(Click image for a larger view)
This interface allows user to query known protein-drug interaction available in the database, where a set of binding residues are bound to existing drug molecule as annotated in the Drugbank database. User may query via several terms: (i) PDB ID (e.g. '1mxd'), (ii) PDB Ligand ID (e.g 'acr'), (iii) Drug name (e.g. 'acarbose') or DrugBank ID (e.g. 'DB00284'), and (iv) Keywords through different categories including drug indication, source organism, macromolecule name and Pfam annotation.
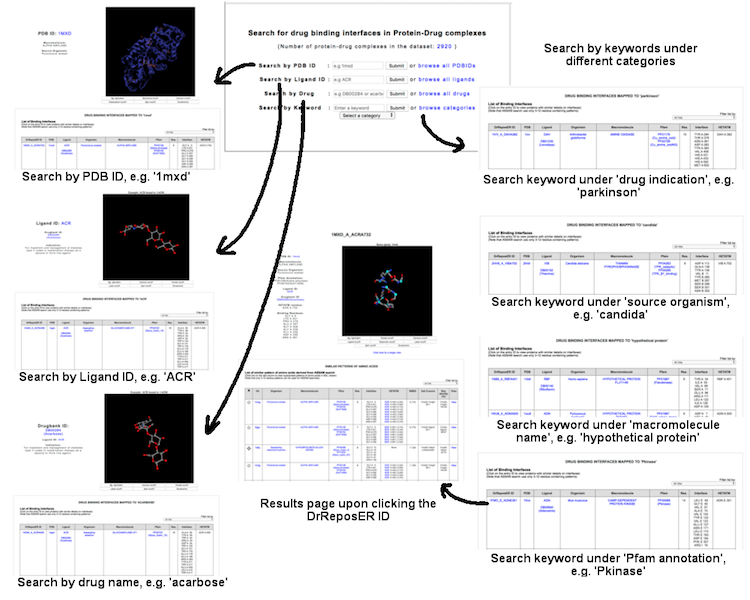
(Click image for a larger view)
Individual binding interface is denoted by the DrReposER ID, represented by '[PDBID]_[PDB Chain ID from which the drug molecule belongs to]_[HETATM record mapped to the drug molecule]'. The first column (DrReposER ID) from the list of hits provides a link that will open a new page describing each entry. The page displays (i) structural details of the PDB, (ii) visualization of the crystal molecule and binding interface, and (iii) a list of similar pattern of amino acids derived from ASSAM searches.
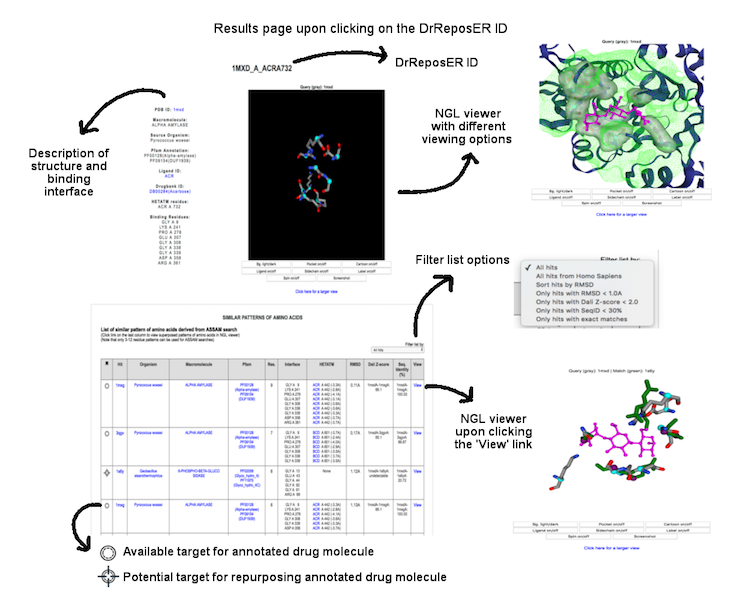
(Click image for a larger view)
This interface allow user to search a query protein against a database of 3D motifs mapped to drug binding patterns. The search is based on SPRITE program (Nadzirin et al., 2012). User may query a PDB ID (four-letter code, e.g. '4cha') or upload a PDB-formatted structure to know whether the protein contain potential 3D motif.
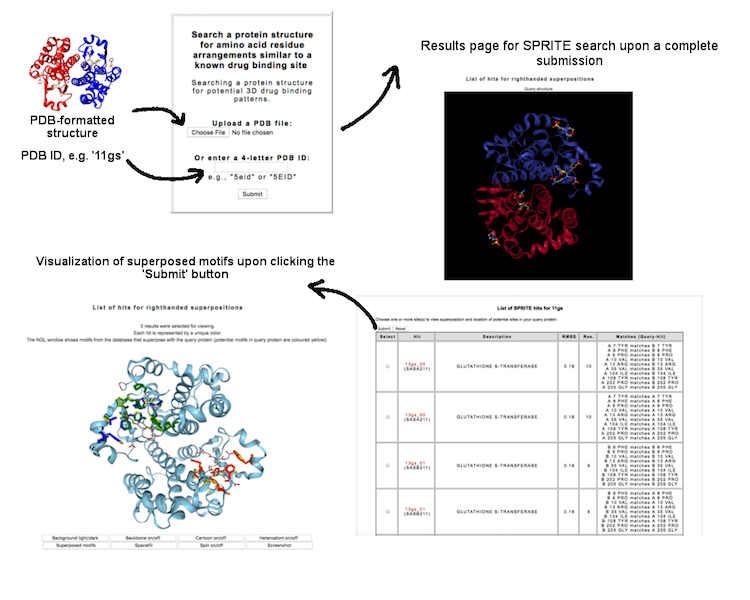
(Click image for a larger view)
Each entry and search results can be visualized using NGL viewer. The user can view superposed patterns of amino acids and ligands through different display options.
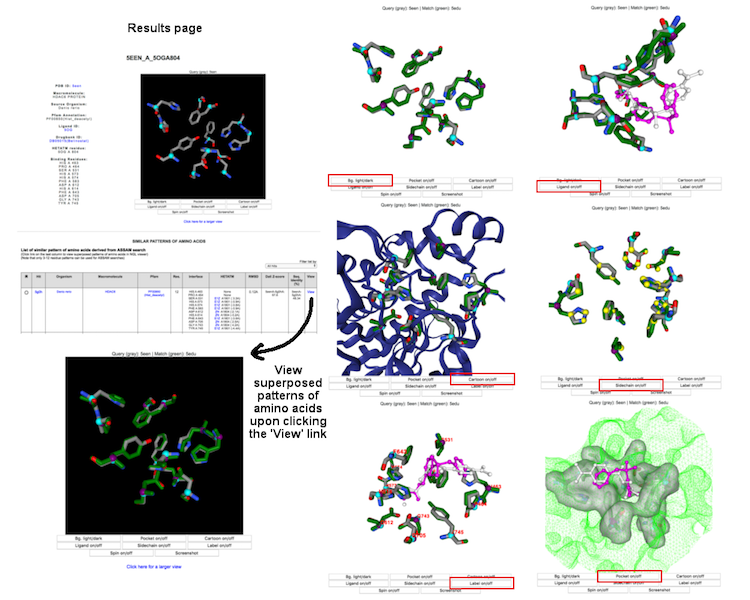
(Click image for a larger view)
| OS | Version | Chrome | Firefox | Microsoft Edge | Safari |
|---|---|---|---|---|---|
| Linux | CentOS 7 | 71.0 | 61.0 | n/a | n/a |
| MacOS | Mojave | 71.0 | 61.0 | n/a | 12.0 |
| Windows | 10.0 | 73.0 | 38.0 | 42.17134.1.0 | n/a |
| Android | 8.0 | 73.0 | 66.0 | n/a | n/a |
| iOS | 12.2 | n/a | n/a | n/a | 5.2 |